Better Health, Brighter Future
A new light on Duchenne muscular dystrophy therapy: Genome editing using lipid nanoparticles
T-CiRA’s Science Series
Duchenne muscular dystrophy is an intractable disease in which muscle atrophy begins during early childhood and subsequently progresses. It is caused by a mutation of the dystrophin gene. Therapies have been developed over the past few years using new technologies to restore the function of this gene, including nucleic acid medicine, gene therapy and genome editing. T-CiRA has developed a novel therapy in which the tools necessary for gene editing are loaded into proprietary lipid nanoparticles and delivered directly to skeletal muscle cells. This overcomes the weaknesses of conventional therapies, and experiments with mice have shown that the effects last for a year.
Radical therapy to restore the dystrophin gene
Muscular dystrophy, which is designated by the government as an intractable disease, causes progressive muscle damage throughout the body. Duchenne muscular dystrophy (DMD), which is caused by mutation of the dystrophin gene, is the most severe form of the disease and affects large numbers of patients.
The dystrophin gene is located on the X chromosome, so DMD occurs only in boys, with an incidence rate of 1 in 3,500 baby boys. As a result of the gene mutation, patients are either completely or partially unable to manufacture dystrophin, a protein needed to maintain muscle structure. Symptoms such as abnormal gait and increased falls appear during early childhood, eventually progressing to difficultly in moving the body and deterioration of respiratory and cardiac functions. Although symptomatic treatments have gradually increased the average life expectancy, few patients survive beyond the age of 30 years at present.
A radical therapy that can restore the function of the gene so that the body is able to produce dystrophin is keenly anticipated. The use of nucleic acid medicine for therapies was approved in Japan in 2020, and clinical trials of gene therapies using viral vectors are currently underway. Research and development of therapies that make use of genome editing is also progressing.
Using proprietary lipid nanoparticles to overcome the problems
Unfortunately, these novel therapies all have various problems for the patient. In the case of nucleic acid medicine, patients must receive an intravenous infusion once a week for the rest of their lives because nucleic acids have a short half-life and disappear quickly in the body. With gene therapy, a viral vector is loaded with the dystrophin gene and introduced into mutated muscle cells to produce dystrophin, but once the gene has been administered, it cannot be re-administered because of the body’s immune response to the viral vector. Genome editing therapy is under development in which a viral vector is used to introduce the tools necessary for genome editing (enzymes, etc. which will be described later) directly into muscle cells, but the therapy is limited to a single administration for the same reason.
T-CiRA has been searching for a way to overcome these problems in order to develop a safe and effective way to treat DMD. Specifically, we have been aiming to deliver the necessary tools to the muscle cells without the use of viral vectors that cause immune responses in genome editing therapies.
The research group has developed a lipid nanoparticle (LNP) made up of four different lipid components (Fig. 1) as a novel delivery method. If they are loaded with the tools needed for genome editing, they will release these tools in the acidic intracellular environment when they reach the target cells. The tools will then be able to perform their required functions.
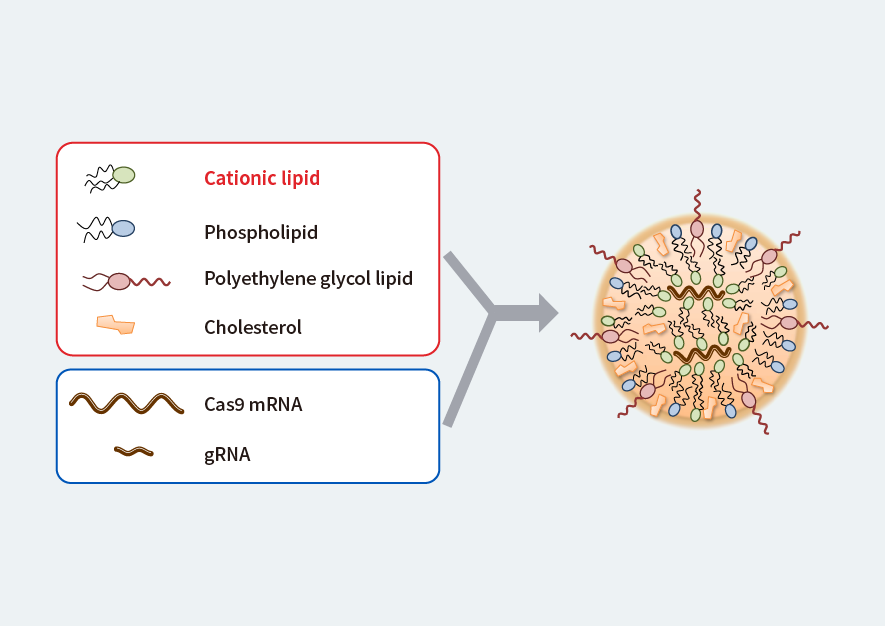
Figure 1. Proprietary lipid nanoparticle (LNP)
The tools needed for genome editing (Cas9 mRNA and gRNA) are loaded into the particles, which are delivered to muscle cells.
Modified from Fig. 1a, Kenjo, E., Hozumi, H., Makita, Y., et al. Low immunogenicity of LNP allows repeated administrations of CRISPR-Cas9 mRNA into skeletal muscle in mice. Nat Commun 12, 7101 (2021). https://doi.org/10.1038/s41467-021-26714-w
DMD therapy by genome editing
What is the mechanism of DMD occurrence? The mRNA produced by the dystrophin gene consists of 79 joined exons (parts that code the information for synthesizing a protein), which is the basis for manufacturing dystrophin. However, some of the exons are missing or duplicated in DMD patients, so that the exons are unable to join up properly, and dystrophin cannot be produced.
The idea of “exon skipping” was developed to address this. Improperly connected exons are skipped (removed) so that the exons before and after them can join up, allowing the production of dystrophin. The dystrophin produced in this way is somewhat shorter than regular dystrophin, but it retains the function of maintaining muscle structure so it may be expected to be effective in reducing symptoms.
In the treatment of DMD by genome editing, exon skipping in the mRNA is brought about by cutting away the parts of the dystrophin gene that correspond to the problem exons. Genome editing is a method for cutting away the target part of a gene, and the tools needed for this are the mRNA for the degrading enzyme Cas9 and guide RNA (gRNA). The gRNA recognizes the part of the dystrophin gene that needs to be cut away, and Cas9 slices the gene at the required sites.
Novel genome editing therapy with lasting effects
There are a number of different types of mutation of the dystrophin gene that cause DMD. One of these is loss of exon 44 (the 44th exon), which causes improper joining between exons 43 and 45, with the result that dystrophin cannot be produced. In this case, if exon 45 is skipped, exons 43 and 46 join well, and dystrophin can be produced.
The research group created a mouse model of DMD that lacks exon 44 and carried out gene editing therapy experiments using the LNP that they had developed (Fig. 2). LNPs were loaded with mRNA for Cas9 and gRNA to recognize the site on the gene before exon 45 and a different gRNA to recognize the site after exon 45. The idea was to send the LNPs to the muscle cells where they could remove exon 45 by slicing the gene before and after this exon.

Figure 2. Genome-editing therapy experiment using LNPs
(a) With DMD, the part of the gene corresponding to exon 44 is lacking, with the result that exons do not join properly in the mRNA, and dystrophin cannot be produced. (b) Model mice with this defect are injected with LNPs variously containing Cas9 mRNA and two types of gRNA for genome editing to take place. The dystrophin gene is cut before and after the part corresponding to exon 45, so that in the mRNA exon 45 is skipped, exon 43 joins to exon 46, and dystrophin can be produced.
Modified from Fig. 2b and Fig. 4a, Kenjo, E., Hozumi, H., Makita, Y., et al. Low immunogenicity of LNP allows repeated administrations of CRISPR-Cas9 mRNA into skeletal muscle in mice. Nat Commun 12, 7101 (2021). https://doi.org/10.1038/s41467-021-26714-w
The results of the experiments were just as planned: exon skipping occurred, and dystrophin was produced. Furthermore, it was confirmed that production of dystrophin was maintained for at least a year with a single administration. This contrasts with the effects of nucleic acid drugs, which only lasted for about a month following administration. In addition, repeated administration is possible with LNPs, and the effect was found to accumulate. In addition, we found that, if the LNPs are injected into ligated blood vessels rather than directly into muscles, they have an effect on a wider range of skeletal muscles.
We intend to continue our research and development by examining the effects in animal models with longer life spans and by improving the LNPs so that they have a greater range of effects. With LNPs, we aim to perfect a safe and reliable treatment for DMD in which the body can produce dystrophin simply through regular injections.